Análise crítica de estudo sobre judicialização de medicamentos raros, revelando vieses, omissões do SUS e dilemas éticos ignorados na gestão da saúde coletiva e bioética brasileira
Análise crítica sobre como decisões do SUS podem abrir espaço para captura privada na triagem neonatal e terapias gênicas. Riscos, incentivos e governança.
Análise crítica sobre como decisões do SUS podem abrir espaço para captura privada na triagem neonatal e terapias gênicas. Riscos, incentivos e governança.
Novas políticas de saúde global, como VBHC e IRA, redefinem o preço de medicamentos órfãos. André Medici analisa o impacto no acesso e sustentabilidade do Brasil.
FAQ explica como a Lei 14.874/2024 altera regras éticas da pesquisa em saúde, reduz direitos dos participantes e ameaça a governança democrática do SUS.
Submeti contribuições à consulta pública: falta trilha para raras/ultrarraras, MEAs, RWE e pesos de severidade. Proponho caminhos para decisões previsíveis e não ao sabor das marés
Submeti contribuições à consulta pública: falta trilha para raras/ultrarraras, MEAs, RWE e pesos de severidade. Proponho caminhos para decisões previsíveis.
Anvisa investiga efeitos adversos graves com base na RDC 406/2020, exigindo notificação em 15 dias e podendo impor ações regulatórias para proteger a saúde.
Jovem com Distrofia Muscular de Duchenne morre após tratamento com Elevidys, gerando preocupações sobre segurança e impacto regulatório da terapia gênica.
“O mercado de pacientes especialistas revela conflitos éticos e desigualdades, transformando participação em saúde em mercadoria e limitando o impacto transformador.”
A proteção patentária é crucial para inovação, mas eleva o custo dos medicamentos órfãos, criando dilemas entre acesso e lucro, como no caso do Claritin.
O STF discute a aprovação do Elevidys, medicamento para DMD que, apesar do alto custo, demonstrou eficácia limitada em ensaios clínicos, segundo a revista Nature Medicine.
Entenda passo a passo os critérios definidos pelo STF para a concessão judicial de medicamentos não listados no SUS. Saiba quais são os requisitos e implicações.
Descubra os novos parâmetros do STF para o fornecimento de medicamentos pelo SUS, incluindo a criação de uma plataforma nacional e a gestão de demandas.
O relatório “NICE Enough?” do Office of Health Economics revela a influente marca do NICE nas decisões de saúde globais. 🌐
As descobertas do estudo enfatizam a reverberação das decisões do NICE além das fronteiras do Reino Unido, moldando a saúde pública em diversos países. Isso ressalta a importância da análise crítica e da adaptação dessas influências nos contextos nacionais. 🚀
Nosso país tem um Legislativo comandado por três bancadas: Bala, Boi e Bíblia (BBB).
Ao redor destes temas gravitam majoritariamente os interesses dos nobres deputados e senadores, que disputam quase à tapa verbas parlamentares para agradar suas bases eleitorais e chantageiam o governo federal em busca de cargos no Executivo.
A interação entre políticas globais de saúde e interesses corporativos não apenas molda a resposta a crises sanitárias, mas também desafia a autonomia de organizações internacionais, como a OMS, ao abordar questões críticas de saúde pública
Não é preciso ser economista para entender a lei da oferta e procura: quanto maior o preço, menor a chance de compra. Ela também vale para o setor farmacêutico,
As associações de pacientes devem lidar com a indústria farmacêutica com ética e transparência. Mas isso não é trivial. Há riscos de conflito de interesses
A Avaliação de Tecnologia em Saúde (ATS) desempenha um papel crucial na avaliação do valor e impacto das intervenções de saúde. No caso das doenças raras, a ATS se torna ainda mais importante devido aos desafios únicos associados a essas condições. Este artigo discute as melhores práticas a serem seguidas ao usar a ATS para deliberar sobre doenças raras, com o objetivo de garantir processos de tomada de decisão justos, transparentes e centrados no paciente. As principais práticas incluem engajamento precoce, contribuição do paciente e especialista, métodos adaptativos de ATS, considerações éticas, modelos inovadores de precificação e reembolso, geração de evidências a longo prazo e colaboração internacional. Ao implementar essas práticas, os processos de ATS podem avaliar efetivamente o valor e o acesso aos tratamentos para doenças raras, melhorando os resultados para os pacientes e abordando as necessidades médicas não atendidas nessas populações.
O acesso às terapias genéticas para doenças raras é um desafio complexo que demanda superação de barreiras significativas. A falta de infraestrutura adequada, modelos de financiamento inovadores e incerteza dos resultados são alguns dos obstáculos a serem enfrentados. No entanto, é fundamental unir esforços e promover ações conjuntas entre diferentes setores para garantir que os sistemas de saúde estejam preparados para oferecer tratamentos acessíveis e de qualidade.
A inovação social farmacêutica refere-se a formas alternativas de pesquisa, desenvolvimento e implantação de medicamentos para doenças raras que se desviam dos caminhos de inovação estabelecidos e se baseiam em princípios de inovação social.
As ATS são cruciais na definição da eficácia, segurança e eficiência de tecnologias médicas. Contudo, pouco se faz para analisar a dinâmica destes colegiados.
Na medida em que não existe consenso sobre que prioridade devemos dar aos doentes mais graves, outras variáveis precisam entrar nessa equação. Destacamos uma.
A Comissão Nacional de Incorporação de Tecnologias no SUS recebe diversas críticas quanto ao seu funcionamento e às suas decisões, sem perspectiva de solução.
Reações belicosas de representantes da entidade diante das críticas razoáveis à sua legitimidade revelam despreparo de agentes públicos no enfrentamento de controvérsias plausíveis.
Do advocacy ao ativismo? O confronto entre associações de pacientes e a indústria farmacêutica (aqui representada pela empresa Vertex Pharmaceuticals) no caso Trikafta®, pode sinalizar uma mudança de fase nas outrora plácidas (e muitas vezes suspeitas) relações entre estes dois atores em solo brasileiro.
Resta saber se tal despertar da consciência cidadã dos pacientes com fibrose cística poderá ser extrapolado para outras associações de pacientes, representando outras enfermidades pouco frequentes. Afinal, preços muitas vezes extorsivos de medicamentos não são problema limitado à fibrose cística.
Há quarenta anos, comemorados no dia 4 de janeiro (quarta-feira), o presidente Ronald Reagan sancionava a Lei dos Medicamentos Órfãos1. Considerada como uma das mais bem-sucedidas leis norte-americanas, ela busca criar incentivos para a produção de medicamentos para o tratamento de doenças raras.
Mas nem tudo é um mar de rosas. Em 2017, uma investigação da Kaiser Health News (KHN) verificou que algumas empresas farmacêuticas estão usando a Lei de Medicamentos Órfãos para criar monopólios e cobrar preços altos por medicamentos que já teriam sido aprovados para uso no mercado de massa nos EUA.
Recentemente, o ministério da saúde anunciou a incorporação ao SUS do Zolgensma, um dos medicamentos mais caros do mundo”: ‘Ouro de tolo’ ou conquista social?
Ela termina após uma primeira prorrogação de prazo e posteriores problemas de acesso à mesma relatados por inúmeras pessoas, sem solução providenciada pela Conitec, apesar de termos levado o problema a representantes da iniciativa Colabore com o Futuro, parceira da referida Comissão.
A discussão a que a consulta pública se refere assume dimensão pública no país em 2015, com a proposição de Projeto de Lei do Senado (PLS) de autoria do então senador Cássio Cunha Lima (PSDB-PB) (PLS 415) que mais tarde viria a dar origem à Lei 14.313 de 21/03/2022.
Este dispositivo legal altera a Lei nº 8.080, de 19 de setembro de 1990 (também conhecida como Lei Orgânica da Saúde), para dispor, segundo seu caput, sobre os processos de incorporação de tecnologias ao Sistema Único de Saúde (SUS) e sobre a utilização, pelo SUS, de medicamentos cuja indicação de uso seja distinta daquela aprovada no registro da Agência Nacional de Vigilância Sanitária (Anvisa).
Não há como se renunciar à criação de um limiar de custo-efetividade explícito para o SUS. No entanto, pelo princípio da precaução, o ônus da prova da conveniência de sua adoção caberia à Conitec.
A edição do Fantástico de domingo passado (4/7) retratou em profundidade os escândalos(clique no vídeo neste link para ter um retrato amplo da situação) envolvendo tanto a empresa Davati (com uma suposta oferta de vacinas da Astrazeneca), como a vacina Covaxin, da Bharat Biontech.
No centro do escândalo relacionado a esta última aparece, em destaque, como principal suspeito, o nome do ex-ministro da Saúde, Ricardo Barros (Progressistas-PR), atual líder do governo na Câmara.
Barros é um velho conhecido dos doentes raros, mas por razões nada lisonjeiras.
Ao longo de todos estes anos pesquisando doenças raras no país, adquiri uma visão privilegiada sobre o modo como administradores públicos procuram confiscar os direitos das pessoas que delas padecem.
Diante das clamorosas lesões a seus direitos fundamentais, não resta aos doentes raros alternativa, senão recorrer ao Judiciário. Não bastassem suas mazelas, suas incapacidades, seu estresse psicossocial, sua doença grave, ainda têm que brigar na justiça para reaver direitos que jamais deveriam ser-lhes negados.
“A saúde é direito de todos e dever do Estado”
Art. 196, Constituição Federal, 1988
Um expediente comum na Administração Pública é, por exemplo, invocar a “reserva do possível” para recusar a estes pacientes o acesso a medicamentos de alto custo.
Ela era ‘arroz de festa’ nas palestras presenciais de gestores informando aos doentes raros que não iriam ter moleza ao reivindicar seus direitos fundamentais.
Adiar, adiar, adiar. Até que você morra. E muitos já morreram, esperando decisões dos tribunais.
O princípio da “reserva do possível” aplicado ao Direito Sanitário vem sendo derrubado nos tribunais com frequência.
Independente disso, alguns administradores públicos com cargos executivos ainda insistem em alardear estas baboseiras diante de platéias de doentes raros, seus familiares e cuidadores.
Como Academia de Pacientes procura trazer aos doentes raros o melhor conhecimento para que lutem por seus direitos, chegou a hora de desmascarar esta falácia.
[su_dropcap style=”flat”]R[/su_dropcap]ecentemente, publicamos um post neste blog que teve muitas visualizações. Por isso voltamos ao assunto. O post anterior buscava …
[su_dropcap style=”flat”]C[/su_dropcap]ausou-me estranheza a declaração do deputado Diego Garcia (Podemos-PR), coordenador da Frente Parlamentar das Doenças Raras, em entrevista …
[su_dropcap style=”flat”]H[/su_dropcap]á poucas semanas, trouxemos para vocêuma grande notícia a respeito das radicais mudanças que se desenham no horizonte em relação ao National Institute for Health and Care Excellence (NICE), que supostamente inspira (é o que dizem) a nossa CONITEC.
Se você não leu as matérias anteriores clique nos links abaixo:
Agora, passamos a destacar um novo aspecto das mudanças propostas e de interesse para você, doente raro, ou familiar de uma pessoa vivendo com doença rara.
[su_dropcap style=”flat”]H[/su_dropcap]á poucos dias trouxemos para você uma grande notícia a respeito das radicais mudanças que se desenham no horizonte …
[su_dropcap style=”flat”]V[/su_dropcap]ocê pode não acreditar, dada toda a sinistrose que a pandemia trouxe para nossas vidas, mas tudo indica que o ano de 2021 será muito interessante para as pessoas que vivem com doenças raras.
Ao menos no Reino Unido. Com alguma sorte, também no Brasil e em outras partes do mundo.
Isto porque o National Institute for Healthcare and Clinical Excellence (NICE), da Inglaterra e do País de Gales, está realizando uma profunda modificação de seus métodos de Avaliação de Tecnologias em Saúde (a maior de todas ao longo de sua história), visando trazer mais justiça e equidade a suas recomendações de incorporação destas aos sistema de saúde locais.
E os maiores beneficiários diretos destas mudanças serão os doentes raros, e, claro, toda a sociedade. Continue com a gente que mais abaixo você vai entender por quê.
[su_dropcap]N[/su_dropcap]o post anterior sobre este tema, vimos que a tragédia da talidomida motivou, em grande parte, a edição de uma emenda à lei em vigor desde 1938 nos EUA, que regulava a produção de medicamentos. Vimos também que esta emenda gerou involuntariamente o que ficou conhecido como “medicamentos órfãos”
A Emenda Kefauver-Harris teve por finalidade garantir a eficácia e segurança dos fármacos. Esta emenda afetou a forma como os medicamentos eram produzidos e a tomada de decisões da parte da indústria farmacêutica sobre que medicamentos produzir ou não. Os medicamentos órfãos representavam assim aqueles que a indústria não tinha mais interesse em produzir, dadas as novas regras motivadas pela publicação da emenda.
[su_dropcap style=”flat”]A[/su_dropcap]s conquistas da nova genética permitiram o nascimento de novas identidades e grupos sociais. Os portadores de doenças raras ajudaram a cunhar o termo “biossocialidade”.
Curiosamente, foi inspirado em uma associação francesa de pacientes com distrofia muscular que Rabinow cunhou este termo.
Na primeira metade da década de 90, Paul Rabinow identificara, na França, a organização de coletivos sociais de nova espécie. Viu ali a inédita construção de identidades individuais e grupais, bem como de práticas, possibilitadas pela descoberta de novas técnicas de diagnóstico genéticas e do monitoramento de riscos e suscetibilidades, em processo que denominou “biossocialidade”.
No futuro existirão grupos formados em torno do cromossomo 17, locus 16, sitio 654.376 alelo com substituição de uma guanina (Paul Rabinow, 1996)
[su_dropcap style=”flat”]A[/su_dropcap] expressão “doenças raras”, tal como a concebemos hoje, surge como consequência de toda a polêmica envolvendo a publicação, nos Estados Unidos, na década de 1960, de uma medida legal: a Emenda Kefauver-Harris ou Emenda da Eficácia dos Medicamentos.
Datada de 1962, ela alterou a Lei Federal de Alimentos, Medicamentos e Cosméticos (Federal Food, Drug and Cosmetic Act), publicada originalmente em 1938.
A Kefauver-Harris foi responsável pela reestruturação completa da forma pela qual os medicamentos eram aprovados nos Estados Unidos (e depois em todo o mundo), afetando até mesmo decisões a respeito de quais deles desenvolver. Foi um dos mais importantes acontecimentos a definir a evolução do cenário regulatório-industrial dos Estados Unidos, desde a década de 1930. A nossa ANVISA, criada em 1990, é fruto destas mesmas preocupações de Estado relativas à segurança e eficácia de medicamentos.
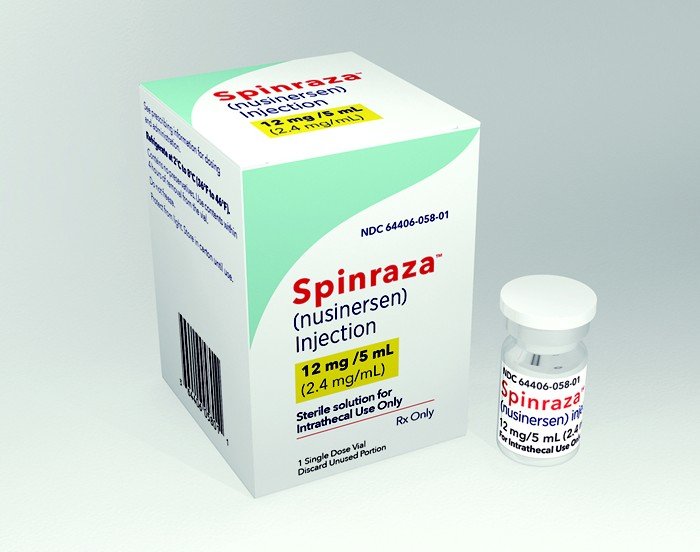
Você leu ontem aqui neste blog que a Conitec emitiu recomendação desfavorável para incorporação de nusinersena (Spinraza) ao SUS nos casos de Atrofia Muscular Espinhal (AME) tipo II e tipo III e que irá encaminhar sua decisão para consulta pública.
Na prática, a prevalecer tal resultado, o medicamento continua incoporado ao SUS somente para os casos de tipo I.
Este é mais um capítulo de um imbroglio que se arrasta desde 2018. A AME é doença neuromuscular de origem genética recessiva rara, com incidência de 1:10.000 nascidos vivos e prevalência estimada de 1-2:100.000 pessoas. Ainda não foi divulgado o Relatório de Recomendação desfavorável e nem as gravações do Plenário que culminou com tal decisão.
Com a decisão divulgada anteontem, somente pacientes com AME tipo I recebem nusinersena pelo SUS. Os restantes a obtém através de judicialização.
Resolvemos realizar uma breve pesquisa para saber como outros países do mundo se posicionaram com relação à incorporação de nusinersena a seus sistemas públicos de saúde. Verifica-se que, em linhas gerais, não considerando os países que ainda estão com negociações de preço em andamento, somente algumas províncias do Canadá restringem o acesso ao medicamento nos mesmos moldes que o Brasil.
No quadro abaixo, quando você ler “Acesso reembolsado”, entenda como se o medicamento estivesse incorporado ao sistema público de saúde.
Os países relacionados abaixo e que constam como concedendo acesso através de Managed Entry Agreement são aqueles que optaram por fazê-lo mediante um acordo de compartilhamento de riscos (como fora proposto para o caso brasileiro para AME tipos II e III) ou assemelhados.
Você deve já ter se perguntado: Como surgem as tais “doenças raras”? Sempre existiram? São invenção da indústria farmacêutica? De todo modo, penso que você deve ter curiosidade de saber como isso tudo começou, pois grande parte de sua vida, como já deve ter percebido, é fortemente determinada por estas duas palavrinhas quase inocentes.
Então vamos começar nossa viagem através desse conceito. Em primeiro lugar, você poderia pensar que “doenças raras” é sinônimo de “doenças pouco frequentes” ou “afecções pouco comuns”.
Na realidade, atualmente, é um conceito um pouco mais preciso que estes, mas ainda vago. Isso porque no Brasil, por exemplo, é qualquer doença que acometa até 65 a cada 100 mil indivíduos, ou até 1,3 pessoas a cada 2 mil. Na Europa, é aquela que atinge uma pessoa a cada 2 mil. Já, nos EUA, é qualquer doença que afete menos de 200 mil pessoas.
Para começo de conversa, você precisa saber que, antes de meados da década de 1970, ninguém poderia imaginar que milhares de patologias totalmente distintas (hoje algo entre 6 mil a 8 mil) poderiam ser agrupadas em uma categoria comum (“doenças raras”) somente tendo a baixa frequência a lhes unir.
Quem depende do Sistema Único de Saúde , tendo uma doença rara ou sendo familiar de uma pessoa vivendo com ela, tende a desconhecer a cultura política que plasmou o SUS na década de 1980. Por isso talvez não compreenda parte importante dos problemas que enfrenta quando precisa recorrer a ele, nesta condição.
Na verdade, o SUS é um dos poucos sistemas de saúde do mundo que aceitou o desafio de oferecer cobertura universal em saúde para mais de 100 milhões de habitantes. Em alguns pontos é bem-sucedido neste intento (como na atenção básica), mas em outros talvez deixe a desejar, como no setor de alta complexidade, que em algum momento será demandado pelos portadores de doenças raras que não possam arcar com os custos dos planos de saúde.
[su_dropcap]N[/su_dropcap]ossas informações divulgadas aqui no blog Academia de Pacientes começam a gerar movimentações importantes e interessantes.
Mas, a comissão de incorporação de tecnologias ao SUS têm divulgado, na pessoa de seus representantes, ou prepostos mais vocais, em apresentações públicas, a ideia de que estaria mentindo quem afirmasse que as Análises de Custo-efetividade (ACE) seriam determinantes nas avaliações feitas por aquela comissão, como costuma se alegar.
[su_dropcap style=”flat”]R[/su_dropcap]ecentemente publicamos no blog Academia de Pacientes informações sobre artigoque acaba de ser publicado no Jornal Brasileiro de Economia da Saúde que revelou que havia “limitações metodológicas relevantes” na qualidade da maioria dos estudos de custo-efetividade realizados para medicamentos contra o câncer, recomendados para incorporação pela Comissão Nacional de Incorporação de Tecnologias no SUS (Conitec). Apesar de os autores terem concentrado sua análise na questão dos medicamentos para tratamento do câncer, achei interessante dividir seus achados com nossas leitoras e leitores, em um blog voltado para doenças raras.
Convidamos então uma de suas autoras a nos detalhar mais um pouco os seus achados. É bom esclarecer que, na entrevista, Tatiane Bonfim Ribeiro falou do lugar de uma pessoa que atua no campo da Economia da Saúde. E não das Ciências Sociais, como costuma ser a perspectiva que adoto em meus comentários por aqui.
Nesse sentido, a entrevista é bastante esclarecedora porque nos permite compreender como estes profissionais refletem sobre as questões de acesso a medicamentos de alto custo e suas avaliações econômicas. As perguntas foram feitas por email, com perguntas fechadas previamente estruturadas. Agradecemos à Tatiane pelo tempo que dedicou a esta tarefa visando esclarecer melhor os pacientes sobre estes processos.
Tatiane é consultora em Avaliação de Tecnologias em Saúde (ATS), especialista em revisão sistemática. Mestre em Saúde Coletiva pela Faculdade de Medicina da USP e Doutoranda em Epidemiologia pela USP
[su_note]Agora queria te fazer um pedido, caro(a) internauta: Se tiver alguma dúvida ou não entender alguma passagem desta entrevista manda sua pergunta nos comentários. “Saber é poder” e conhecer estas coisas é fundamental para que você possa exercer sua cidadania com mais conhecimento de causa.[/su_note]
Tatiane defende mais transparência em todo o processo
Academia de Pacientes: Quais as implicações dos seus achados no estudo, para os pacientes, a sociedade e para a qualidade dos gastos em saúde? O que lhe chamou mais a atenção?
Tatiane: O principal objetivo do trabalho foi avaliar a qualidade dos estudos econômicos (análise de custo-efetividade – ACE) submetidos a Conitec e com recomendação favorável para incorporação no SUS. Dos 10 medicamentos oncológicos incorporados no período, somente cinco tinham avaliação de custo-efetividade disponível, sendo este o objeto da nossa análise. Nossos resultados mostram que a maioria apresentou qualidade baixa e moderada, o que implica em estudos econômicos com incertezas que podem dificultar a tomada de decisão por parte da Conitec. Os modelos utilizados nas ACE são cheios de pressupostos, pois estes fazem estimativas de um cenário hipotético com base em probabilidades e dados extrapolados para a realidade local.
Sabemos que os estudos econômicos, como de custo-efetividade e impacto orçamentário, compõem uma parte das análises em Avaliação de Tecnologias em Saúde (ATS) que primeiramente avalia a efetividade e segurança comparada entre a nova intervenção (p. ex. novo medicamento para câncer de mama) e as opções terapêuticas já existentes. Com o aumento dos gastos em saúde, as análises econômicas podem influenciar diretamente nas decisões de recomendação pela Conitec e eficiência do sistema de saúde, desta forma, estudos com determinadas limitações e omissão de algumas análises (como análises de sensibilidade) produzem resultados incertos. Por fim, algumas limitações podem diminuir a confiabilidade do estudo, podendo até invalidar os resultados devido a pressupostos incorretos imputados no modelo. Como a Conitec utiliza estes dados para subsidiar suas decisões, as limitações dos modelos podem impactar diretamente no acesso à nova tecnologias em saúde.
[su_dropcap style=”flat”]N[/su_dropcap]o artigo “O ‘remédio mais caro do mundo’ e os dilemas para o SUS e o STF”, publicado nesta Folha no dia 26 de agosto, os autores Daniel Wei Liang Wang, Luís Correia, Adriano Massuda e Ana Carolina Morozowski propõem um debate amplo com a sociedade sobre o preço do Zolgensma. O medicamento é indicado para o tratamento da atrofia muscular espinhal (AME), uma doença rara que causa paralisia muscular progressiva. Penso que começar pelo preço é uma habilidosa maneira de encerrar essa discussão.
Aldous Huxley dizia que “às vezes é necessário restabelecer o óbvio”. Não custa lembrar que Zolgensma está no rol das “terapias gênicas curativas”. É, portanto, dotado de forte componente de inovação disruptiva, com potencial de curar (algo nada trivial). É resultante da “revolução biotecnológica”, iniciada em meados da década de 1970.
[su_dropcap]O[/su_dropcap] leitor tem acompanhado aqui o julgamento que prevê a formulação de tese de repercussão geral (Tema 6) relativa ao dever do Estado de fornecer medicamento de alto custo a portador de doença grave que não possui condições financeiras para comprá-lo e quando não estiverem previstos na relação do Programa de Dispensação de Medicamentos em Caráter Excepcional, do SUS. A votação estava prevista para se encerrar hoje (28/8), mas com o pedido de vista ela fica adiada.
[su_dropcap]T[/su_dropcap]al como fizemos em matéria recente sobre o tema, vamos destrinchar o acontecimento em um FAQ (ou um pergunta-e-resposta) para que você possa entender o que está em jogo em sua vida, especialmente se você precisa de medicamentos de alto custo para tratamento de doenças raras.
Como você viu ontem aqui, o julgamento no Supremo Tribunal Federal (STF) foi suspenso porque o Ministro Gilmar Mendes pediu vistas ao Plenário. A decisão então fica adiada.
O que estava sendo julgado até ontem no STF?
Trata-se de decisão que fixará a Tese de Repercussão Geral do Tema 6. Ela definirá quando pacientes terão o direito de receber tratamentos não incorporados pelo SUS.
[su_box title=”O que é o Tema 6?” style=”soft” box_color=”#2215d5″ title_color=”#f0ce48″ radius=”5″]Dever do Estado de fornecer medicamento de alto custo a portador de doença grave que não possui condições financeiras para comprá-lo.[/su_box]
Artigo atualizado em 23/08/2020. às 15h31 [su_note]Polichinelo é uma antiga personagem-tipo e burlesca da commedia dell’arte, cujas raízes remontam ao …
[su_dropcap style=”flat”]N[/su_dropcap]a terça-feira passada (4/8), a Conitec divulgou em seu site a intenção de realizar uma pesquisa. Ela visaria “identificar usuários do SUS e associações de pacientes existentes no Brasil e consolidar um banco de dados para ações de participação social que serão realizadas pela Secretaria-Executiva da Comissão”. A pesquisa consiste em um questionário de 33 perguntas, sobre o qual falaremos mais abaixo.
Para justificar a realização da mesma, a nota informa que “é uma das frentes de trabalho da Conitec promover ações que fortaleçam a participação social no processo de incorporação de tecnologias em saúde no SUS”.
[su_dropcap style=”flat”]A[/su_dropcap] Comissão Européia encomendou um estudo cujos resultados, ainda inéditos, sugerem que a maioria dos medicamentos órfãos teriam sido lançados ainda que não existissem incentivos específicos para a sua produção, como a concessão de exclusividade de mercado por 10 anos.
Há casos de medicamentos com 20 anos de exclusividade de mercado, como o milionário eculizumabe, comercializado sob a marca Soliris®. É como se descobríssemos, de repente, que aquele menino ófão chorão, supostamente desamparado e descapitalizado pelo infortúnio, fosse herdeiro de uma grande fortuna. O assunto mereceu destaque na prestigiosa British Medical Journal.
Desta forma a indústria aproveita-se indevidamente de um incentivo que, buscando corrigir supostas distorções de mercado, acaba provocando outras: lucros astronômicos dos laboratórios com produtos que haviam recebido incentivos para sua produção por seu alegado baixo retorno comercial.
[su_dropcap style=”flat”]S[/su_dropcap]istemas públicos de saúde do mundo inteiro, como o SUS, estão sendo cada vez mais pressionados a entregar, em tempo hábil, medicamentos novos e frequentemente de alto custo para seus cidadãos.
Isso inclui medicamentos para o tratamento de doenças raras, que afetam um pequeno número de pacientes, muitas vezes graves (que ameaçam a vida e/ou causam uma incapacidade significativa), adquiridas geneticamente e que apresentam seus primeiros sintomas na infância.
[su_note]Atenção: toda vez que você ler a abreviatura TDR no texto entenda como “tratamento para doenças raras (e ultrarraras)” [/su_note]
Em muitas partes do mundo, incentivos financeiros e processos inovadores para facilitar a pesquisa, o desenvolvimento e a comercialização foram implementados/autorizados para tratamentos de doenças raras (TDR). Tudo isto visando reduzir a expressiva necessidade não-atendida de tratamentos modificadores destas doenças.
Na União Européia, considera-se que essas medidas foram bem-sucedidas, com 176 medicamentos autorizados até este ano pela Agência Européia de Medicamentos (EMA).
Decisões relativas ao reembolso, preço e disponibilidade de tratamentos nos sistemas de saúde são frequentemente informadas por Avaliações de Tecnologias em Saúde (ATS), que são baseadas em evidências internacionais, analisadas por um comitê deliberativo de especialistas (ou não) para determinar o benefício clínico adicional e/ou a relação custo-benefício das novas tecnologias em um sistema de saúde específico. No Brasil, como você já deve saber, este papel é desempenhado pela Conitec.
Em 9 de novembro de 1962, a já extinta revista Life estampou em suas páginas uma matéria impactante, de autoria de Shana Alexander.
Ela retratava o Admissions and Policy Committee (Comitê de Admissões e Política) do Centro de Hemodiálise de Seattle. O Comitê fora criado naquele ano para selecionar as poucas pessoas que seriam admitidas na nova e minúscula unidade de hemodiálise criada pelo Dr. Belding Scribner, o inventor do shunt arteriovenoso.
O comitê era composto por sete membros anônimos: um pastor, um advogado, um empresário, uma dona de casa, um líder sindical e dois médicos.
Alguém mais espirituoso o apelidara de “Comitê de Deus”.
[su_dropcap style=”flat”]O[/su_dropcap] Instituto Unidos pela Vida promoveu, na terça-feira passada (21/7), no âmbito de seus bate-papos sobre Fibrose Cística. uma live com a participação de técnicas da Conitec, para entender melhor como seus pacientes (e, por extensão, a sociedade em geral) podem participar das consultas públicas daquela comissão, em processos de incorporação de medicamentos e outras tecnologias de saúde ao SUS.
Com a diretora da organização, Verônica Stasiak, como moderadora, o evento teve a Conitec representada por Fabiana Raynal Floriano e Bruna Viana. Coincidentemente, a live aconteceu no mesmo dia de outra que já destacamos aqui no blog , com a participação do ex-secretário da SCTIE, Denizar Vianna.
A escolha do tema dessa nova live foi oportuna. Afinal, os pacientes de fibrose cística viram há poucas semanas dois de seus medicamentos mais promissores terem sua recomendação ao SUS desaconselhada. Decisão que os pacientes agora pretendem tentar reverter na consulta pública sobre Ivacaftor (Kalydeco®) e Ivacaftor + Lumacaftor (Orkambi®), ambos do laboratório Vertex.
O vídeo é bem esclarecedor e reina nele atmosfera de civilidade, o que é algo sempre desejável. Fabiana e Bruna respondem às perguntas a elas endereçadas por Verônica e pelos internautas. As respostas podem ser úteis a quem quer que se interesse por participar de consultas públicas futuras, acerca de seus medicamentos de predileção. Recomendo que assistam.
[su_youtube_advanced url=”https://youtu.be/-Hs8GlmB0fk” rel=”no” title=”Live – Tire suas dúvidas sobre consultas públicas na Conitec”]
Há um momento que me foi especialmente prazeroso no vídeo.
Com informações e análises independentes, podemos exercer pressão específica para que os tratamentos experimentais sejam tratados adequadamente. Até o momento, houve pouca pressão, porque contamos com especialistas para interpretar para nós o que está acontecendo. Eles nos dizem só o que não lhes vai criar problemas. As empresas que querem seus lucros, os burocratas que querem seu território e os médicos que querem evitar causar polêmica estão todos à mesa. As pessoas com AIDS que querem suas vidas devem estar lá também.
(John James, ativista e editor do Aids Treatment News, 1986).
Basear-se somente em instituições oficiais para nossa informação é uma espécie de suicídio coletivo.
(John James, 1986)
A luta contra a Aids foi o primeiro movimento social nos EUA a realizar a conversão em massa de “vítimas” da doença em pacientes-especialistas. Fizeram isso por conta própria, como autodidatas. Muitas vezes se posicionando até mesmo contra os laboratórios.
Entenda algo sobre isso, vendo o trailer abaixo.
[su_youtube_advanced url=”https://youtu.be/aKHq84W_oJs” rel=”no” title=”120 batimentos por minuto”]
[su_dropcap style=”flat”]N[/su_dropcap]o momento em que a Comissão Européia trabalha para concluir a elaboração de seu futuro Plano de Combate ao Câncer, a Eurordis, maior coalizão européia de associações de pacientes com doenças raras, empenha-se para que o documento contemple adequadamente as necessidades das pessoas afetadas por cânceres raros. O Brasil é terra-de-ninguém quando o assunto são cânceres raros.
O Brasil não possui dados oficiais sobre a incidência e/ou prevalência destes cânceres na população geral e sequer adota esta nomenclatura em sua atuação governamental. Eles também não estão contemplados na política nacional de doenças raras. Mas, se transportarmos para o Brasil os dados europeus, em termos absolutos, podemos estimar que o país conta com 1,4 milhão de pessoas vivendo com cânceres raros.
São números impressionantes. Câncer raro, na União Européia (UE), é aquele que acomete até seis pessoas a cada 100 mil habitantes por ano. Estima-se que existam 5,1 milhões de pessoas com um câncer raro na Europa, entre 200 tipos de tumores catalogados segundo a RareCareNet. De acordo com a Rare Cancers Europe, 24% de todos os casos de câncer, incluindo os pediátricos, são raros. Segundo a Eurordis, um em cada cinco novos pacientes com diagnóstico de câncer possui um câncer raro. Os cânceres raros contribuem com 20 a 30% dos novos casos de câncer nos Estados-membros da União Européia, estimando-se em 650 mil o número anual de novos diagnósticos de cânceres raros na UE.
Há quem acredite que, no futuro próximo, todos os cânceres serão raros. Não por sua baixa ocorrência, mas graças à crescente compreensão dos mecanismos fisiopatológicos a eles subjacentes, o que acaba contribuindo para a divisão de categorias mais amplas de tumores em entidades patológicas menores e mais bem definidas. Pavimenta-se assim o caminho para a medicina personalizada, visto com antipatia por alguns profissionais da saúde coletiva.
[su_dropcap style=”flat”]N[/su_dropcap]o dia 11 de março deste ano, o Plenário do Supremo Tribunal Federal (STF) decidiu que o Estado não é obrigado a fornecer medicamentos de alto custo solicitados judicialmente, quando não estiverem previstos na relação do Programa de Dispensação de Medicamentos em Caráter Excepcional, do Sistema Único de Saúde (SUS). As situações excepcionais ainda serão definidas na formulação da tese de repercussão geral (Tema 6) (1). A decisão, tomada no julgamento do Recurso Extraordinário (RE) 566471, atinge mais de 42 mil processos sobre mesmo tema.
Para Marcus Dantas, a decisão final a ser adotada pelo STF sobre o fornecimento de medicamentos, se for restritiva, pode ter consequências graves para muitas pessoas. Em especial, para aqueles pacientes que obtiveram liminares e decisões favoráveis de juízes e tribunais para receber e iniciar seu tratamento, mas seus processos ainda estão em andamento (ou foram sobrestados aguardando a decisão com repercussão geral). Em outras palavras, na avaliação do paciente e servidor público, todos aqueles processos sem decisão definitiva (ou seja, a grande maioria).
[su_youtube_advanced url=”https://www.youtube.com/watch?v=29hE_ONQ8bE” rel=”no” title=”Entidade quer incluir pessoas com deficiência em grupo de risco para COVID-19″] [su_dropcap style=”flat”]N[/su_dropcap]ão se fala …
PRISCILA PEREIRA MENINO [su_dropcap style=”flat”]N[/su_dropcap]uma prática pouco usual, a ANVISA impôs sigilo sobre a decisão tomada em relação ao caso …
Artigo de Priscila Pereira Menino [su_dropcap style=”flat”]A[/su_dropcap] Agência Nacional de Vigilância Sanitária (Anvisa) foi criada, em 1999, para evitar que …
Caixa-preta é presente de grego! / Foto de PublicDomainPictures
[su_dropcap style=”flat” size=”5″]N[/su_dropcap]a semana passada você leu aqui que a CONITEC divulgou relatório de recomendação preliminar, desaconselhando a incorporação do riociguate ao SUS, e que isto teria suscitado protestos de associações de pacientes.
Alega-se que os processos de tomada de decisão daquela Comissão seriam pouco transparentes. E que transparência neste domínio tornaria estes processos mais justos.
Este tipo de reação da sociedade não é privilégio do Brasil. Outras agências de Avaliação de Tecnologias em Saúde (ATS) espalhadas pelo mundo também são criticadas, quando suas decisões desagradam pacientes e seus familiares. O prazo da Consulta Pública sobre a incorporação do Riociguate foi prorrogado [su_highlight background=”#d5f427″]até o dia 16 de janeiro[/su_highlight].
Lucio Luzzatto e colaboradores
Publicado originalmente na revista The Lancet (20.7.2018)
Contam-se nos dedos os casos de uma única medida legislativa que tenha mudado tão radicalmente a política industrial na indústria farmacêutica como a Lei de Medicamentos Órfãos, assinada nos EUA em 1983.
A Lei foi elaborada para facilitar o desenvolvimento de medicamentos para doenças raras e outras condições de saúde (1) e os incentivos propiciados pela Lei resultaram na aprovação de 575 medicamentos e produtos biológicos para doenças raras entre 1983 e 2017 pela Food and Drug Administration (agência que controla medicamentos e alimentos nos EUA) (2). Um verdadeiro sucesso. Em 2000, a Comissão Europeia aprovou legislação semelhante para medicamentos órfãos (MOs). De fato, as doenças, e não os medicamentos, são órfãs porque todas estes medicamentos são muito caros, (3) nota destoante nesta história de sucesso .
O Conselho Nacional do Ministério Público (CNMP) realizou nos dias 29 e 30 de novembro de 2018, em Brasília/DF, o Seminário Reflexões sobre a Judicialização da Saúde: um diálogo interinstitucional. O blog AcadP esteve presente e traz um resumo dos temas que interessam diretamente às pessoas vivendo com doenças raras e seus familiares. A programação pode ser consultada aqui. A realização do evento coube à Comissão Extraordinária da Saúde (CES) e à Unidade Nacional de Capacitação do Ministério Público (UNCMP).
O evento teve como principal finalidade “promover o diálogo entre as instituições públicas envolvidas na judicialização da saúde: Ministérios Públicos Federal, do Trabalho, de Contas e Estaduais, Poder Judiciário, Conselho Nacional de Justiça, Defensoria Pública, Procuradorias do Estado, Ministério da Saúde, Conselhos Federais de Medicina e de Farmácia, Conselhos de Saúde e Gestores de Saúde”.
No centro dos debates, a necessidade de “racionalização da judicialização”. O tema da ‘racionalização’ é amplamente estudado no campo das Ciências Sociais (e tende a ser bem traiçoeiro para doenças raras), e sobre ele trataremos em futuros posts. Segundo os organizadores, esta seria “prioridade em função do elevado e crescente número de ações judiciais propostas no país anualmente para a obtenção de medicamentos, procedimentos, tratamentos médicos, produtos e tecnologias em saúde”.
Um caso recente envolvendo a Comissão Nacional de Incorporação de Tecnologias ao SUS (CONITEC) e o Instituto Oncoguia pode servir de inspiração para que inúmeros portadores de doenças raras (e frequentes), bem como seus familiares, questionem na Justiça os relatórios de recomendação daquela comissão, com decisões pela não-incorporação de determinadas tecnologias de saúde ao SUS.
Explico. Recentemente o Instituto Oncoguia, após avaliar o relatório de recomendação da Conitec na Consulta Pública CONITEC nº 47/18, sobre a proposta de incorporação dos medicamentos indicados para o tratamento de carcinoma de células renais, identificou que a Conitec emitiu um tipo de consideração que não era da sua alçada, extrapolando assim sua missão institucional, sem definir-se pela recomendação ou não de incorporação de determinados medicamentos.
Spinraza (nusinersen) é a primeira e até o momento única medicação para Atrofia Muscular Espinhal, esta frequentemente fatal doença genética. A despeito de sua aprovação no ano passado, o NICE recuou por conta de deliberações sobre a melhor maneira de gerenciar o medicamento.
O ponto nevrálgico tem sido seu preço de 75 mil libras (cerca de 368 mil reais) por dose, excluindo impostos. Spinraza deve ser administrado em quatro doses a cada duas semanas , seguidas de doses de manutenção a cada quatro meses. Com este preço, o custo anual do tratamento é de 450 mil libras (cerca de 2,2 milhões de reais) para o primeiro ano e 225 mil libras (1,1 milhão de reais) para os anos seguintes.
Um estudo descritivo e detalhado, conduzido por pesquisadoras do Departamento de Medicina Preventiva da Universidade de São Paulo (USP) , encontrou um grande descompasso entre os relatórios de recomendação de tecnologias divulgados pela CONITEC e o que a lei determina como o que deve ser observado pela mesma, no que se refere a suas atribuições legais.
É importante ressaltar que o estudo em questão não tratou especificamente dos pareceres da Comissão relativos a medicamentos órfãos, indicados para o tratamento e prevenção de doenças raras. Dados os resultados apresentados no referido artigo, podemos imaginar que, em se tratando de doenças raras, os descompassos da CONITEC sejam ainda maiores.
A Agência Européia de Medicamentos é o organismo responsável por centralizar o procedimento de autorização de medicamentos a nível europeu. Depois de ser autorizado na EMA, cada novo medicamento deve seguir procedimentos de licenciamento nacionais, que estão ligados a precificação, reembolso e Avaliações de Tecnologias em Saúde (HTA). Passar por todas estas fases exige tempo; em alguns países da União Européia até mesmo uns 12 anos: esta longa espera representa uma sentença de morte para pessoas vivendo com cânceres raros e outras doenças raras e ultrarraras.
Há cerca de quatro anos, a EMA lançou um novo projeto sobre Caminhos Adaptativos ou Adaptive Pathways (AP), em inglês, para aprovação condicional de medicamentos. Este projeto despertou debates acalorados nos Estados-Membros da UE, entre pacientes, cientistas, profissionais, pesquisadores, gestores em saúde e indústria farmacêutica. Vamos tentar entender melhor este projeto e suas consequências imediatas.
Como já vimos falando aqui neste blog, em diversos posts, os métodos empregados pela Conitec para deliberar sobre incorporação de medicamentos órfãos ao SUS são totalmente desaconselhados em sistemas de saúde que optaram por verdadeiramente contemplar as necessidades de assistência terapêutica e farmacêutica de doentes raros. A literatura especializada tem comprovado isso fartamente.
Este emprego inadequado de Avaliações de Tecnologias em Saúde (ATS) convencionais é uma das razões prováveis para a judicialização galopante que (supostamente) incomoda bastante os gestores de saúde e (verdadeiramente) doentes raros e seus familiares. Também pode explicar o fato de a Conitec em seus quase sete anos de existência só ter recomendado a incorporação de uma meia dúzia de medicamentos órfãos ao SUS.
Mas o que é a Análise de Decisão Multicritério (ADMC)? Nada mais é do que uma ATS ampliada, razão pela qual não haveria, a nosso ver, maiores problemas para a Conitec empregá-la, se houvesse vontade política. Mas vamos a uma definição mais formal da ADMC:
Um conjunto de métodos e abordagens de auxílio à tomada de decisão, onde as decisões são baseadas em mais de um critério, o que evidencia o impacto, sobre a decisão, de todos os critérios aplicados e a importância relativa a eles atribuída.
Desta forma, abre-se mão de adotar apenas um critério ou outro (como o da custo-efetividade, p. ex.) nas ATS ou, na melhor das hipóteses, tornam-se transparentes e consistentes os demais critérios considerados pelos organismos como a Conitec em seus julgamentos.
Eu conheci Tony, em 2001, quando ele me foi encaminhado com uma severa fraqueza muscular. Confinado a uma cadeira de rodas e incapaz de cuidar de si mesmo, ele recebera o diagnóstico de esclerose lateral amiotrófica (ELA), um distúrbio neurodegenerativo fatal. Verificou-se que Tony na verdade tinha síndrome miastênica de Lambert-Eaton, um distúrbio neuromuscular raro, mas tratável. Depois de iniciar o tratamento com a 3, 4 diaminopiridina (3,4 DAP), ele recuperou grande parte de sua força muscular e, de modo importante, sua capacidade de andar e cuidar de si.
Tiago Farina Matos, diretor jurídico do Instituto Oncoguia, tem realizado uma interessante campanha informal, reivindicando que a Conitec dê acesso irrestrito, seja virtual ou presencial, a suas reuniões plenárias para pacientes, vinculados a associações ou não. Quer maior transparência na tomada de decisões desta Comissão. O blog Academia de Pacientes resolveu aceitar o desafio de Tiago e verificar como a mais respeitada agência de Avaliações de Tecnologias em Saúde (ATS) do mundo lida com o assunto.
Em post recente em seu perfil no Facebook, Tiago informa que em sua 61ª Reunião, realizada em novembro de 2017, os membros do Plenário da Conitec “concordaram em só manter o acesso de alguns poucos convidados e autorizados a apenas uma pequena parte da reunião, a parte em que o solicitante da incorporação apresenta o seu ponto de vista”. Na avaliação de Tiago, do modo como deliberou a Conitec na reunião, “feita a apresentação (protocolar), todos os participantes da sociedade civil devem sair da reunião para o que os 13 membros possam discutir o assunto a portas fechadas”.
Quanto à participação de usuários ou representações de usuários no plenário da CONITEC, concordou-se que ela pode continuar sendo feita por meio de convite da Secretaria-Executiva da Comissão. Nos casos em que o pedido partir dos usuários ou representações, conforme já ocorre, a participação dessas pessoas poderá ser autorizada como ouvinte, e exclusivamente, no momento da apresentação do tema de interesse.
Pois bem, como o National Institute for Health and Clinical Excellence (NICE) lida com estas questões?
Doenças infecciosas são um grave problema de saúde pública no mundo inteiro, causando inúmeras complicações sérias, incluindo distúrbios autoimunes em …
Você não precisa ser muito inteligente para imaginar que os desafios lançados a sistemas públicos de saúde (especialmente aqueles de caráter universal, como o SUS) por medicamentos novos e de alto custo não são privilégio do Brasil. Todos os países do mundo que, de alguma forma, ousaram enfrentar o problema representado pelas doenças raras defrontam-se, em maior ou menor grau, com a questão do preço destas novas tecnologias em saúde.
A diferença entre eles e o Brasil é que aqueles buscaram soluções para resolver tais impasses e garantir, em algum grau, a assistência integral à saúde a pessoas que vivem com doenças raras. O Brasil ainda engatinha nesse campo.
A solução que aqui mostraremos tem sido adotada por inúmeros países da Europa e por Estados Unidos, Nova Zelândia, Austrália, Canadá, dentre outros. Apresenta seus desafios (como acordar pela manhã também os têm). Mas, segundo analistas, são plenamente factíveis em realidades como a do SUS, desde que se tomem as cautelas usuais, visando o sucesso de políticas públicas. Para eles, é mera questão de tempo a possibilidade de serem adotados no Brasil.
Os genes nas células de seu corpo desempenham um importante papel na sua saúde. De fato, gene(s) defeituoso(s) pode(m) fazer você adoecer.
Cientes disso, pesquisadores têm trabalhado durante décadas em formas de modificar ou substituir genes defeituosos por outros saudáveis para tratar, curar ou prevenir uma doença ou condição clínica.
Este é um achado robusto da literatura internacional sobre o qual a Conitec jamais se pronunciou: Não é recomendável empregar Avaliações de Tecnologias em Saúde de modo convencional, quando o assunto são medicamentos para doenças raras.
Diante dos dados que nos são permitidos conhecer a respeito desta Comissão, podemos afirmar (salvo melhor entendimento) que ela aplica metodologias convencionais em Avaliação de Tecnologias em Saúde (ATS) para decidir sobre a incorporação deste ou daquele medicamento ao SUS, indistintamente, seja este órfão ou convencional.
Por que medicamentos órfãos precisam de uma ATS diferente?
As atuais metodologias para ATS de medicamentos convencionais são principalmente baseadas em dois tipos de critérios: a eficácia clínica (efetividade) e a custo-efetividade de novos fármacos, comparados a outros produtos no mesmo campo terapêutico (no caso de existirem).
Embora os métodos-padrão de ATS tenham um importante papel na otimização da eficiência da provisão de cuidados em saúde em geral, estes tendem a resultar em acesso restrito e desigual a novos medicamentos órfãos por parte dos portadores de doenças raras, por diversas razões, incluindo as seguintes:
A imprensa falada, escrita e televisada tem divulgado o desespero de doentes raros e seus familiares que, de uma hora para outra, há alguns meses, estão sem receber medicamentos a eles assegurados por ordem judicial. Recusar cumprir ordem judicial costumava ser crime, em governos legítimos e democráticos. Mas este não é o tema do post aqui.
A esta altura, você já sabe o que é a CONITEC e o que ela faz. Não se pode dizer que a CONITEC seja fraca em propaganda. Se não souber ainda, clique aqui.
Mas vamos refrescar sua memória: Criada em 2011, por meio de Lei Federal e regulamentada por Decreto Presidencial, a Conitec estabeleceu novas regras para incorporação de tecnologias em saúde no SUS. Através desta Lei Federal (Lei 12.401) a Avaliação de Tecnologias em Saúde (ATS) foi institucionalizada como “critério indispensável para tomada de decisão sobre a incorporação de tecnologias em saúde no SUS”.
Resumindo bem, ATS é uma avaliação sistemática de propriedades, efeitos e/ou impactos de tecnologias de saúde. Seu principal propósito é auxiliar tomadores de decisão na definição de políticas de saúde. É parte de um movimento que ficou conhecido como Medicina Baseada em Evidências, que também já abordamos neste blog.
A Conitec realizou, nos dias 19 e 20 de outubro, no Hotel Transamérica Prime International Plaza (SP), o fórum “Entendendo a Incorporação de tecnologias em saúde”. Promovido pelo Departamento de Gestão e Incorporação de Tecnologias em Saúde do Ministério da Saúde (DGITS/MS), em parceria com o Hospital Alemão Oswaldo Cruz, o evento “teve como proposta levar informação e educação a representantes de pacientes, no intuito de ampliar e qualificar o engajamento social nas etapas que antecedem a disponibilização de tecnologias em saúde no SUS”, segundo informa comunicado, em tom apologético, publicado na página da instituição.
A nota prossegue informando que “durante o evento, 103 participantes receberam informações sobre como a CONITEC trabalha, sobre os critérios considerados na incorporação de tecnologias e na elaboração de protocolos clínicos e sobre os canais existentes para envolvimento no processo”.
Em post anterior, revelamos para você que todo o arcabouço da assistência farmacêutica contemporânea no Brasil está fortemente escorado no que se convencionou chamar “medicina baseada em evidências” (MBE). Um dos mais prestigiados instrumentos da MBE é a “avaliação de tecnologias em saúde” (ATS). No Brasil, a obrigação legal de fornecê-las é da CONITEC. Não é o caso de nos alongarmos aqui, mas você precisa entender que as ATS convencionais não são instrumento adequado para contemplar os direitos dos doentes raros à saúde, o que inclui o medicamento.
Mas o que são as ATS? Em tese, poderíamos dizer que são uma forma de pesquisa que visa avaliar questões médicas, éticas, econômicas e sociais relacionadas ao uso de uma determinada tecnologia em saúde (um medicamento, um procedimento cirúrgico, um instrumento, um procedimento, um protocolo clínico etc.). Por que “em tese”? Porque na prática cada país pode escolher enfatizar um dos critérios, em detrimento de outros. No Brasil, por exemplo, tudo leva a crer que os critérios mais valorizados são os econômicos (e não os éticos, médicos ou sociais), salvo melhor juízo, no que se refere a medicamento voltados para doenças raras. Por isso você tem que entrar na justiça pra receber medicamento de alto custo.
Você deve ter visto! Um novo tratamento para o câncer recentemente aprovado pela FDA (a agência que controla drogas e medicamentos nos EUA) custa 475 mil dólares (cerca de um 1,5 milhão de reais). Tirando um ou outro tratamento para doenças ultrarraras, trata-se da terapia mais dispendiosa já concebida. O caso pode representar uma interessante lição para quem depende de medicamentos para doenças raras para sobreviver.
A indicação é para um grupo pequeno de pacientes com limitadas ou quase inexistentes opções de tratamento. Foi aprovada para leucemia linfoblástica aguda pediátrica refratária (resistente a outros medicamentos) ou com pelo menos duas recidivas (recaídas). A cada ano, surgem aproximadamente 3 mil novos casos , mas a maioria pode ser tratada com abordagens convencionais. Sobram, assim, cerca de 600 pacientes candidatos ao tratamento, segundo a fabricante.
De uma perspectiva de negócios (sim, internauta, fazer negócios não é pecado!), isso não é uma grande quantidade de consumidores finais. Assim, parte da razão para seu alto preço (assim como para outros medicamentos para doenças raras) é a necessidade de recuperar os custos de produção.
Mas ainda que isso possa parecer uma cifra assombrosa, analistas financeiros garantem que ela é bastante razoável.
Com divulgação prévia discreta, foi realizada na quinta feira (24/8), audiência pública na Comissão de Assuntos Sociais do Senado para instruir o Projeto de Lei do Senado nº 415, de 2015, (PLS) de autoria do Senador Cássio Cunha Lima (PSDB-PB).
O relato do que ali se passou você pode ler aqui e neste outro link. Abaixo, alguma contextualização para seu benefício.
O PLS em questão propõe alterar a Lei 8.080, de 19 de setembro de 1990, também conhecida como “Lei Orgânica da Saúde”. Se aprovada, a proposta tornará obrigatória a definição e divulgação pública de um limiar de custo efetividade (RCEI) (entenda este termo, mais abaixo) para balizar as ações da Comissão Nacional de Incorporação de Tecnologias ao SUS (Conitec) e a distribuição aleatória dos processos de incorporação de tecnologias em saúde (medicamentos, dentre outras) aos Nucleos de Avaliação de Tecnologias em Saúde. A audiência pública foi requerida pela senadora Ana Amélia (PP-RS).
Entender o que se passou na audiência pública é da maior importância para todas aquelas pessoas que dependem de medicamentos órfãos ou de alto custo, ou têm recorrido à Justiça para obtê-los.
Você não deve saber, mas Jack Klugman, falecido em dezembro de 2012, teve papel importantíssimo na aprovação de uma lei (link em inglês) nos Estados Unidos que possibilitou a produção de fármacos que, não fosse isto, não seriam de interesse comercial para a indústria farmacêutica. Como protagonista do seriado médico Quincy, M.E., na ocasião, pôde fazer a diferença conquistando corações e mentes para a causa.
Nós usamos cookies para otimizar nosso blog e nossos serviços.
Funcional
Sempre ativo
O armazenamento ou acesso técnico é estritamente necessário para a finalidade legítima de permitir a utilização de um serviço específico explicitamente solicitado pelo assinante ou utilizador, ou para a única finalidade de efetuar a transmissão de uma comunicação através de uma rede de comunicações eletrónicas.
Preferências
O armazenamento ou acesso técnico é necessário para o propósito legítimo de armazenar preferências que não são solicitadas pelo assinante ou usuário.
Estatísticas
O armazenamento ou acesso técnico que é usado exclusivamente para fins estatísticos.O armazenamento técnico ou acesso que é usado exclusivamente para fins estatísticos anônimos. Sem uma intimação, conformidade voluntária por parte de seu provedor de serviços de Internet ou registros adicionais de terceiros, as informações armazenadas ou recuperadas apenas para esse fim geralmente não podem ser usadas para identificá-lo.
Marketing
O armazenamento ou acesso técnico é necessário para criar perfis de usuário para enviar publicidade ou para rastrear o usuário em um site ou em vários sites para fins de marketing semelhantes.
Utilizamos cookies para melhorar a sua experiência no nosso site. Ao utilizar o nosso site, você concorda com os cookies.
Este site usa cookies
Os sites armazenam cookies para melhorar a funcionalidade e personalizar sua experiência. Você pode gerenciar suas preferências, mas bloquear alguns cookies pode afetar o desempenho e os serviços do site.
Cookies essenciais permitem funções básicas e são necessários para o bom funcionamento do site.
Name
Description
Duration
Cookie Preferences
This cookie is used to store the user's cookie consent preferences.
30 days
These cookies are needed for adding comments on this website.
Name
Description
Duration
comment_author
Used to track the user across multiple sessions.
Session
comment_author_email
Used to track the user across multiple sessions.
Session
comment_author_url
Used to track the user across multiple sessions.
Session
Google reCAPTCHA helps protect websites from spam and abuse by verifying user interactions through challenges.
Name
Description
Duration
_GRECAPTCHA
Google reCAPTCHA sets a necessary cookie (_GRECAPTCHA) when executed for the purpose of providing its risk analysis.
179 days
Google Tag Manager simplifies the management of marketing tags on your website without code changes.
Name
Description
Duration
cookiePreferences
Registers cookie preferences of a user
2 years
td
Registers statistical data on users' behaviour on the website. Used for internal analytics by the website operator.
session
Os cookies de estatísticas coletam informações anonimamente. Essas informações nos ajudam a entender como os visitantes utilizam nosso site.
Google Analytics is a powerful tool that tracks and analyzes website traffic for informed marketing decisions.
Contains information related to marketing campaigns of the user. These are shared with Google AdWords / Google Ads when the Google Ads and Google Analytics accounts are linked together.
90 days
__utma
ID used to identify users and sessions
2 years after last activity
__utmt
Used to monitor number of Google Analytics server requests
10 minutes
__utmb
Used to distinguish new sessions and visits. This cookie is set when the GA.js javascript library is loaded and there is no existing __utmb cookie. The cookie is updated every time data is sent to the Google Analytics server.
30 minutes after last activity
__utmc
Used only with old Urchin versions of Google Analytics and not with GA.js. Was used to distinguish between new sessions and visits at the end of a session.
End of session (browser)
__utmz
Contains information about the traffic source or campaign that directed user to the website. The cookie is set when the GA.js javascript is loaded and updated when data is sent to the Google Anaytics server
6 months after last activity
__utmv
Contains custom information set by the web developer via the _setCustomVar method in Google Analytics. This cookie is updated every time new data is sent to the Google Analytics server.
2 years after last activity
__utmx
Used to determine whether a user is included in an A / B or Multivariate test.
18 months
_ga
ID used to identify users
2 years
_gali
Used by Google Analytics to determine which links on a page are being clicked
30 seconds
_ga_
ID used to identify users
2 years
_gid
ID used to identify users for 24 hours after last activity
24 hours
_gat
Used to monitor number of Google Analytics server requests when using Google Tag Manager